Seenivasan Hariharan, Post Doctoral Researcher, University of Amsterdam | Quantum Chemistry with Quantum Computers | Computational Chemistry
Quantum computing, a new technology, is causing a storm in the well-established computing industry. Quantum computing is considered a disruptive technology that comes with an advantage over classical/traditional computing in ‘certain’ areas that can be directly relevant to society. Some of the potential applications of quantum computing include, acceleration of drug development, identification of green materials for batteries in cars, identification of better catalysts for fertilizer production, optimization of the supply chain, portfolio optimization, improved financial modelling, better weather forecasting and climate change predictions, etc. The past five years have seen a tremendous increase in the number of start-ups, and the amount of public and private funding in the field of quantum computing. Rapid developments in quantum hardware, quantum algorithms, and quantum software have also been realized during this period.
Simulation of molecules (many body systems) has been predicted to have a quantum advantage earlier than other fields where quantum computing can be relevant. This article is primarily aimed at quantum computing enthusiasts interested in the applications of quantum algorithms to chemistry. Having said that, I will also attempt to scratch the surface and go beyond what is usually discussed in popular articles. In this article, I will shed light on what quantities are of interest in heterogeneous catalysis – that can help in the identification of better catalysts for fertilizer production, for example. Understanding these quantities can help tune the activity and selectivity of some important catalytic reactions.
Figure AFigure B
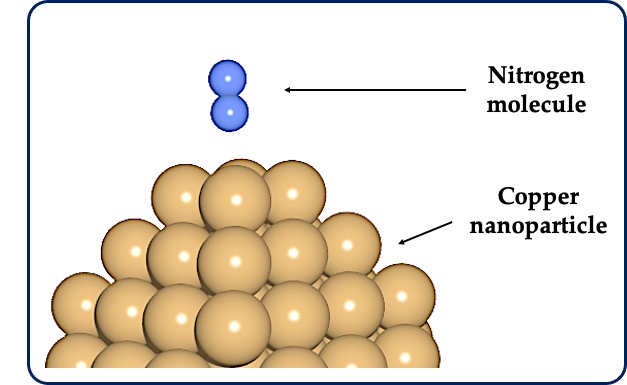
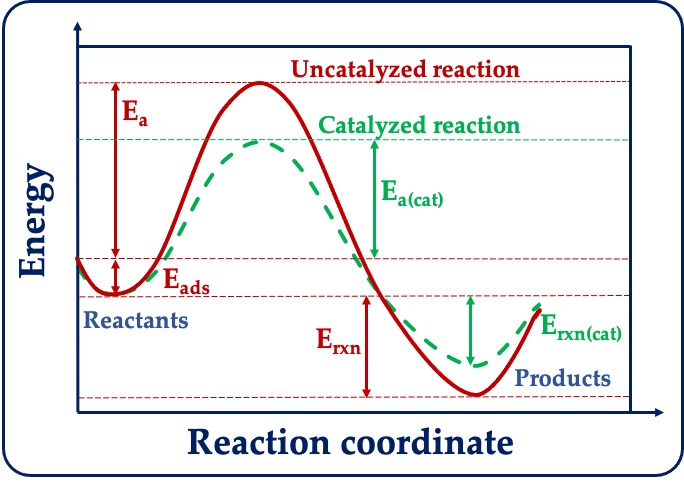
Figure: (A) A typical example of the initial step of a heterogeneous catalytic reaction. Shows adsorption of a nitrogen molecule (N2) in the gas phase on a metal (Cu) nanoparticle in a solid phase. (B) Schematic energy diagram showing the difference between an uncatalyzed and catalyzed reaction. Three relative energy quantities, viz., adsorption energy (Eads), activation energy barrier (Ea) and reaction energy (Erxn) are shown for these two types of reactions.
Computational quantum chemistry plays a critical role in some of the examples listed above. For this article, let us consider the example of identifying better catalysts for a sustainable future. Over 80% of the chemicals produced today depend on heterogeneous catalysts. In heterogeneous catalysis, a reaction is accelerated in the presence of a catalyst, which is not consumed during the reaction. It is called heterogeneous because, the phase of the reactants and the catalysts are different from each other. In general, the reactants are either in the gas or liquid phase, while the catalysts are in the solid phase. Catalysts have two very important properties: activity and selectivity. Activity refers to the property of a catalyst to activate a given chemical reaction. Specificity refers to the property of a catalyst to catalyze a specific reaction (breaking a specific bond, for example). The higher the activity and specificity, the better the catalyst. To have control over the activity and selectivity of a catalyst, it is crucial to understand the interaction of the molecules with the catalyst, which is usually made of metal nanoparticles (Figure A). Only when a complete understanding of a chemical reaction is achieved at the atomic level can one try to improve the activity and selectivity of a given chemical reaction.
In heterogeneous catalysis, three quantities, viz., adsorption energy (Eads), activation energy barrier (Ea), and reaction energy (Erxn) – are central to understanding the feasibility of a chemical reaction (Figure B). The first quantity, adsorption energy (Eads), provides information on how strongly or weakly molecules are bound to the nanoparticle. For an ideal catalytic reaction, the adsorption of the reactant molecule should be neither strong nor weak. The second quantity is the activation energy barrier (Ea). As the name suggests, the reactants must cross the barrier for the reaction to occur. This quantity provides information on how fast a reaction can proceed; slow reactions have a larger activation energy barrier. Based on the value of Ea, one can say whether a reaction is kinetically feasible or not. Catalysts play a major role in reducing the barrier due to various electronic and geometrical reasons, thereby accelerating the reaction. The final quantity is reaction energy (Erxn), the difference in energy between reactants and products. This quantity provides information on the thermodynamic feasibility of a reaction. When Erxn is positive, the reaction is known as endothermic (product formation unfavourable) and when Erxn is negative, the reaction is known as exothermic (favourable product formation). It is important to note that the three quantities discussed above are relative energies and not total energies. This means that for understanding chemical reactions, we may not require very accurate total energies but well-converged relative energies. The relative energies should be within “chemical accuracy” to be able to reliably predict the outcome of a reaction.
It is well established that quantum phase estimation (QPE) and variational quantum eigensolver (VQE) are the two major algorithms for calculation of ground state energies for molecules. Because QPE requires a large number of error-corrected qubits to estimate the ground state energy, it is only appropriate for the fault-tolerant era. On the other hand, VQE and its variants are found to be suitable for ground state energies in the noisy-intermediate scale quantum (NISQ) era. Though the NISQ quantum hardware is error prone, it can be useful in the context of heterogeneous catalysis for classically intractable systems, as we are targeting the relative energies rather than the accurate total energies.
Production of ammonia using conventional heterogeneous catalysis is less efficient. This reaction uses high temperatures and pressure and involves some undesirable side reactions. On the other hand, in nature, the nitrogenase enzyme efficiently produces ammonia in a green and sustainable process. Inspired by nature, scientists are investigating the mechanism of nitrogen dissociation on the active site of nitrogenase enzyme. The central active site of this nitrogenase is made up of the iron-molybdenum-sulphur co-factor (FeMoCO), which is surrounded by proteins. However, calculating the energies of the individual steps leading to the mechanism of this reaction is difficult for classical computers due to the size of the molecule and the nature of the interaction in FeMoCO. Owing to the quantum mechanical nature of the electrons, quantum computing is expected to accurately simulate this reaction. While the resource estimates have already been made, it will be time before we know the answer to the burning question of whether quantum computing will be a solution to this problem. Based on the interest in simulating this system using quantum computing, we can safely say that the simulation of FeMoCO is the poster child for the application of quantum computing to quantum chemistry.
While practical computational quantum chemistry has almost always depended on heuristics, it has immensely helped chemists understand the atomic-level mechanisms of complicated chemical reactions over the years. There has been a demand for more accurate methods, and developments have been steady in this direction. Today, computational quantum chemistry plays an integral role in many industries, focusing on pharmaceuticals, catalysis, and novel materials. I strongly believe that quantum computational chemistry is one such method that is powerful and should be added to the arsenal of existing methods to tackle quantum chemistry problems relevant to humankind.
About Seenivasan: Seenivasan Hariharan is a Researcher working on the applications of quantum algorithms for chemistry and materials science applications. Primarily, he uses a variant of the famous variational quantum eigensolver (VQE) algorithm and runs his calculations on simulators and on actual quantum hardware offered by IBM (superconducting qubits).
Seenivasan received his PhD from IISER Kolkata, India, in 2016. During his PhD, he worked on atomistic-scale understanding of elementary chemical reactions at surfaces, relevant to heterogeneous catalysis, using various computational and theoretical methods. Prior to his current position, he was postdoctoral researcher at Texas Tech University, Texas, USA and Leiden Institute of Chemistry, Leiden, The Netherlands, exploring the atomistic-scale phenomena and understanding mechanisms of chemical reactions at various surfaces.
Become a SheQuantum registered user now to access our exclusive educational resources in Quantum Computing from global experts.
Follow and Share – to get the best of quantum computing courses, industry & academic updates, expert interviews, lecture series and educational resources in quantum computing!
Disclaimer: All content are from the respective organisations web page and provided as it is without any changes or modifications. The “Read More” direct the readers to the respective company’s websites. If you want your company related to quantum computing news, press releases and about events, conferences & webinars to be published here in our news letter, please contact us.